
La malattia può manifestarsi in modo molto differente anche in pazienti che hanno un simile danno al DNA
La sindrome da delezione 2q37 è una condizione cromosomica ben definita: le persone con questa patologia perdono una piccola ma variabile quantità di materiale genetico vicino alla fine di uno dei loro due cromosomi 2 e questo influenza il loro sviluppo, ma il modo in cui si manifesta la sindrome può variare molto. I ricercatori, infatti, hanno ripetutamente trovato differenze sorprendenti tra pazienti che hanno perso quasi esattamente lo stesso segmento di DNA. Non sono ancora disponibili dati su incidenza e prevalenza della malattia ma, stando ai casi ad oggi noti, le femmine sono colpite più frequentemente (60% dei casi).
LE BASI DELLA PATOLOGIA: CROMOSOMA 2 E BANDA 2q37
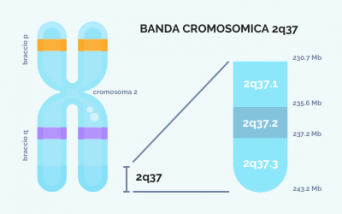
Le cellule umane, ad esclusione di quelle germinali, contengono normalmente 23 coppie di cromosomi, per un totale di 46. Di questi, il cromosoma 2 – anch’esso presente in duplice copia – è uno dei più grandi e contiene più di 1.600 geni. Per vedere meglio questi agglomerati di materiale genetico, non visibili a occhio nudo, si può usare una tecnica che prevede la loro colorazione e l’ingrandimento: a questo punto saranno visibili la struttura, che ricorda una X con 2 bracci più corti (chiamati p) e due più lunghi (chiamati q), e delle bande chiare e scure. In questo modo è possibile evidenziare anche la banda 2q37, dove “q” indica il braccio lungo del cromosoma, “3” indica la regione e “7” indica la banda, che può essere suddivisa in più segmenti.
Ogni banda contiene milioni di copie di basi del DNA: la banda 2q37, localizzata sull’estremità del cromosoma 2, ha poco più di 12 milioni di coppie di basi, corrispondenti a circa 200 geni. Pur essendo un numero elevato, in realtà è molto piccolo se paragonato alla quantità di materiale genetico totale. Purtroppo, non si conosce la funzione di tutti questi geni ed è in fase di studio la loro correlazione con i sintomi associati alla sindrome. L'identificazione dei geni responsabili delle caratteristiche della sindrome da delezione 2q37 è preziosa e può aiutare a guidare studi futuri, ma se un gene presumibilmente responsabile è mancante, non sempre significa che le caratteristiche della malattia associate saranno presenti: altri fattori genetici e ambientali hanno spesso un ruolo nel determinare la presenza o l'assenza di una particolare caratteristica, aumentando la complessità della situazione. Inoltre, in questa sindrome la dimensione della delezione non è standard, ma varia da caso a caso. Il cromosoma, infatti, può rompersi in un qualsiasi punto delle bande chiamate 2q37.1, 2q37.2 o 2q37.3 (vedi immagine) e, di conseguenza, i pazienti possono avere parti di cromosoma mancanti di dimensioni e caratteristiche diverse.
Un gene che è collegato - in modo abbastanza sicuro - a diversi aspetti della sindrome è denominato HDAC4, si trova nel segmento 2q37.3 ed è fondamentale per il corretto sviluppo delle ossa, della cartilagine e del cuore. Agisce anche nella sopravvivenza delle cellule nervose e gioca un ruolo nello sviluppo di disturbi del comportamento, convulsioni e disabilità intellettive. Purtroppo, i fattori che interferiscono con la corretta espressione di HDAC4 non sono stati ancora completamente spiegati.
EREDITARIETÀ E DIAGNOSI
Nella maggior parte dei casi, i bambini con delezione 2q37 nascono da genitori senza alcun danno cromosomico rilevante: se entrambi i genitori hanno cromosomi normali, la mutazione viene definita de novo. Le delezioni 2q37 di solito si verificano all'improvviso e senza una ragione evidente, ma l'unico modo per esserne certi è analizzare i cromosomi di entrambi i genitori. In ogni caso, nessun fattore ambientale, dietetico, lavorativo o di stile di vita dei genitori è noto per causare questi cambiamenti cromosomici nel nascituro. Se i genitori di un bambino con sindrome da delezione 2q37 hanno cromosomi normali, il rischio di avere un altro figlio con la patologia è appena superiore al rischio di chiunque altro. Se invece uno dei due genitori ha un riarrangiamento cromosomico che coinvolge la banda 2q37 - pur senza sintomi - la possibilità di avere altri casi in famiglia aumenta considerevolmente.
La diagnosi della sindrome da delezione 2q37 si basa sull’analisi citogenetica (anche chiamata mappa cromosomica o cariotipo), cioè lo studio dei cromosomi, e sulla caratterizzazione molecolare. La diagnosi genetica prenatale è possibile quando la delezione o il riarrangiamento associato è stato precedentemente identificato in un membro della famiglia. La diagnosi differenziale, cioè quel procedimento che permette di escludere malattie con sintomi sovrapponibili, dovrebbe includere altre patologie da delezione del cromosoma e la sindrome di Prader-Willi. Anche le varie forme di pseudoipotiroidismo con osteodistrofia ereditaria di Albright dovrebbero essere valutate, ma nei pazienti con delezione 2q37 i livelli di calcio, fosforo e ormone paratiroideo sono solitamente nella norma.
SINTOMATOLOGIA
Le persone con una delezione 2q37 mostrano di solito un ritardo nello sviluppo e possono anche avere problemi più o meno gravi di salute e, a volte, di comportamento. Le prime manifestazioni della malattia variano molto: alcuni bambini nascono praticamente sani e mostrano deboli segni della sindrome, come il basso tono muscolare, la difficoltà iniziale ad alimentarsi - che ha come conseguenza un mancato o fluttuante aumento di peso - e alcune caratteristiche insolite del viso o delle mani. Il ritardo nello sviluppo è un primo sintomo comune, di solito associato ad altre manifestazioni, come un severo eczema o delle difficoltà persistenti di alimentazione. Una piccola percentuale di bambini non sta bene già dalla nascita e presenta, ad esempio, difficoltà respiratorie o patologie cardiache o gastrointestinali. Può capitare, anche se occasionalmente, che l’epilessia sia uno dei primi sintomi della sindrome.
Durante la crescita, insieme al ritardo nello sviluppo, si può evidenziare basso tono muscolare e articolazioni lasse, cambiamenti caratteristici nell'aspetto del viso, delle mani e dei piedi, difficoltà di apprendimento, tendenza all’obesità e, a volte, crisi epilettiche e comportamenti assimilabili ai disturbi dello spettro autistico. Altre problematiche rilevate comprendono eczema, asma e frequenti infezioni alle vie respiratorie e alle orecchie. Nel 30% dei pazienti si verificano malformazioni cardiache, gastrointestinali, genitourinarie e del sistema nervoso centrale. I problemi comportamentali sono comuni e possono includere comportamenti ripetitivi, gravi deficit di comunicazione e interazione sociale, movimenti stereotipati, aggressività intermittente, iperattività, disturbo da deficit di attenzione, disturbo ossessivo-compulsivo e disturbi del sonno.
GESTIONE E PROGNOSI
La gestione della sindrome da delezione 2q37 dovrebbe essere multidisciplinare: i pazienti avrebbero bisogno di un attento screening medico tra la nascita e i 5 anni, per via delle eventuali malformazioni associate alla malattia, e di un follow-up in centri di riferimento per persone con disabilità intellettiva. Non è disponibile una terapia ad hoc per la sindrome, ma gli interventi mirati alla gestione dei sintomi aiutano molto, attraverso farmaci specifici, fisioterapia, logopedia, esercizio fisico, dieta e sostegno per l’apprendimento.
La prognosi è molto diversa da paziente a paziente, dato che alcuni presentano una sintomatologia lieve mentre altri hanno gravi malformazioni congenite o disabilità intellettive. La maggior parte dei pazienti in età adulta non è autonoma e alcuni di loro possono sviluppare anche disturbi psichiatrici.
Fonti:
- Orphanet
- Genetic and Rare Diseases Information Center (GARD)
- Understanding Rare Chromosome and Gene Disorders












Seguici sui Social